第22章 心血管病急诊药物临床药理学
第一节 血循环功能不全对药代动力学的影响
一、心脏急症时血循环功能不全的病理生理特点
(一)血循环功能不全 指血循环(主要为心脏)不能提供足够心排血量以满足机体组织代谢需要,它是心脏急症时改变药代动力学最重要和最常见的原因。
(二)心肺复苏 心肺复苏时心排血量明显减少,平均动脉压的下降,在病人大于50%,在实验狗,少于30%。麻醉狗的实验,室颤造成的心脏骤停时,脑血流量减少不明显,心和肾以及其他器官的血流量则明显减少。
二、受影响的药代动力学环节
上述病理生理改变往往涉及三个主要参数(图22-1)。
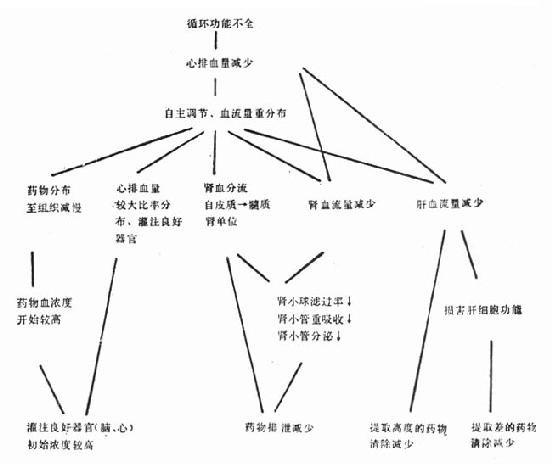
图22-1 循环功能不全对药代动力学的影响
(一)分布容量 心排血量减损,机体血流量重分布,较大部分的心排血量供给脑、心等器官的灌注。心功能不全患者的中心室分布容量较正常减小。因此按通常剂量给予药物常引起非期望的过高血药浓度,出现不良反应。
(二)清除率 药物自血浆的清除,可经肝而生物转化为其他形式(灭活性或活性);或经肾排泄;或经肝及肾两者而消除。决定消除率有两个独立因素:通过器官的血流率和清除机制的活性。这些因素在心功能不全时常降低。
其他因素如体液潴留可增加水溶性药物的分布容量;低蛋白血症可增加蛋白结合率高的药物分布容量,或减少高提取率药物的肝清除率。合用的药物可产生血流动力学效应,从而改变对药物处置。
(三)清除半减期(T[XB]1/2[/XB])大多数药物以一级动力学过程清除,即自血浆移除的药量与当时的血浆药物浓度成比例。分布容量(vd)与清除率(cl)决定清除半减期(T[XB]1/2[/XB]),其关系为:T[XB]1/2[/XB]=vd×0.693/cl,如vd减小,cl成比例也减少,可不改变T[XB]1/2[/XB];利多卡因常先给予负荷量以期加速达到治疗水平,但心衰时vd减小,故易使血浓度较高;cl延缓,T[XB]1/2[/XB]也延长,因而心衰病人利多卡因达到稳态的时间也显著较长,增量时需更加逐步进行。
第二节 各别药物临床药理学
心血管药物种类和制剂众多,本章的讨论包括其药理作用、药代动力学参数、临床应用、适应证、禁忌证、用法、剂量、剂型和注意事项、不良反应及药物交互作用。
| 一、正性肌力药 强心甙 西地兰、毒毛旋花子甙K、地高辛 非强心甙类 多巴胺、多巴酚丁胺、氨吡酮、咪利酮 二、利尿药 呋喃苯胺酸、利尿酸、布美他尼 三、抗心律失常药 利多卡因、奎尼丁、普鲁卡因胺、双异丙吡胺、茚满丙二胺、丙胺苯丙酮、美心律、普萘洛尔、胺碘酮、溴牙苄胺、维拉帕米、阿托品、三磷酸腺苷、苯妥英钠、异丙肾上腺素、钾盐、镁盐 四、扩血管药及其他降压药、抗心绞痛药 硝普钠、酚妥拉明、硝酸甘油、巯甲丙脯酸、降压嗪、利血平 五、抗凝药、溶栓药 肝素、链激酶、尿激酶、组织型血浆素原激活剂 |
{4}一、强心甙(洋地黄,Digitalis)
正性肌力药(强心药)大多属强心甙。目前国内急诊常用的强心甙有毛花甙丙、毒毛旋花子甙K、地高辛。
(一)药理作用 强心甙作用性质类同。
1.正性肌力作用 洋地黄抑制肌膜Na[SB]+[/SB]-K[SB]+[/SB]-ATP酶,增加Na[SB]+[/SB]内流,K[SB]+[/SB]外流,细胞内Na[SB]+[/SB]堆积,促使Na[SB]+[/SB]—Ca[SB]2+[/SB]交换,Ca[SB]2+[/SB]内流增加,改变细胞内[Ca[SB]2+[/SB]]而增加心肌收缩力。强心甙使衰竭心脏的心排血量和心脏作功增加,左心室舒张末压和舒张末容量增加,它对心功能降低者的作用较正常心脏显著。
洋地黄有收缩血管作用。甚至发生在增强心肌收缩作用之前,对人冠状动脉也有类似作用,但仅出现在快速给药时,若给药缓慢则可避免。
2.电生理作用 洋地黄提高迷走神经活性,抑制窦房结,可减慢心率;通过减慢房室结传导速度和延长其有效不应期,可减慢心室反应(房颤时),中毒量强心甙可增高自律性,抑制传导性而产生各种心律失常。
(二)药代学 不同制剂在作用的强弱、快慢、久暂有差别。
西地兰(毛花甙丙,Cedilanid,Lanatoside-C)口服吸收差,仅10%~40%,主要静脉给药。地高辛仅小部分与蛋白结合,主要由肾排泄,80%为原型,17%为主要代谢产物地高辛。T[XB]1/2[/XB]约36h。
地高辛(Digoxin)口服吸收60%~85%,25%与蛋白结合,60%~90%由肾排泄,T[XB]1/2[/XB]为33~36h。肾功能不全时延长。主要口服,也可静脉给药用于急诊。
毒毛旋花子甙K(StrophanthineK)口服几乎不吸收(2%~5%)、90%~100%由肾排泄,T[XB]1/2[/XB]为12~19h。
(三)临床应用 急诊时宜选用静脉给药。
1.主要适应证 ①房颤或房扑,伴室率增快者;②急性心力衰竭或原有心力衰竭加重;③折返性室上性心动过速(首选用维拉帕米,W-P-W综合征时不用)。
用法、剂量、剂型见表22-2。
表22-2 常用速效强心甙:成人剂量与用法
| 制剂 | 剂型(安瓿) | 急症剂量与给药方法 | 生效速度 | 最大作用时间 | 效力维持时限 |
| 毛花甙丙 | 0.4mg(2ml) | 开始0.4mg小壶入,或20ml葡萄糖水稀释后,缓慢静注,必要时2~4h后再给0.2~0.4mg | 10min | 1/2~2h | 1~2d |
| 毒毛旋花甙 K | 0.25mg(1ml) | 开始0.25mg,用20ml葡萄糖水稀释,缓慢静注,必要时2~4h后再给0.125mg | 3~10min | 1h | 1~2d |
| 地高辛 | 0.25mg(1ml) | 开始0.25~0.5mg稀释后静注,4~6h后再给0.25mg | 10min | 14h | 1~2d |
2.禁忌证 ①Ⅱ度或Ⅲ度房室传导阻滞或窦性心动过缓;②肥厚性梗阻性心肌病,洋地黄增加收缩性,可加重梗阻;③预激综合征;④直接电转复有发展严重室性心律失常危险;可能因钾与儿茶酚胺耦联,增加洋地黄毒性。转复前应先停地高辛24h,洋地黄毒甙72h。
(四)不良反应 洋地黄治疗指数低,常需近中毒量的60%以产生治疗效应。以下多种因素可影响机体对洋地黄敏感性,甚至在治疗剂量时也可能发生毒性反应(表22-3)。
| 1.电解质紊乱 | 3.疾病状态 |
| 低血钾 | 甲状腺功能减低 |
| 高血钙 | 低血氧症 |
| 低血镁 | 肾衰竭(地高辛) |
| 高血钠 | 心肌炎 |
| 碱中毒 | 新近心外科手术 |
| 严重心脏疾病 | |
| 2.药物 | 4.老年 |
| 失钾利尿剂 | (减少肌肉质,减少ATP酶结合部位,减少分布容积,减少肾功能使Digoxin排泄减少) |
| 生胃酮(Carbenoxolone) | |
| 皮质激素 | |
| 利血平 | |
| 奎尼丁 | |
| 儿茶酚胺 | |
| 胺碘酮 | |
| 维拉帕米、硝苯吡啶 |
(五)血浓度监测的临床意义 测定稳态洋地黄甙血浓度(地高辛服后6h或下次给药前取血标本),对洋地黄中毒的判断有一定参考价值。治疗病例,通常地高辛血浓度为0.5~2ng/ml,在此范围,一般说血浓度测定对指导治疗无大意义;因血清中的含量不一定可靠地反映心肌内的含量。
(六)用药注意事项 ①给药前应详细了解近两周内使用洋地黄情况,如1周内曾用过洋地黄或不详者,不应按通常给药量,而应将剂量减少和分次给予,以防过量;②急性心肌梗死最初1~2天,避免用洋地黄,因此时期心肌心电不稳,易出现心律失常;③急诊静脉给药后常需换用口服制剂,常用地高辛,在末次西地兰后6h开始,0.125mg q6h,两次,次日起0.25mg,每日1次。
(七)药物交互作用 其他药物(如排钾性利尿药)产生的低钾可增强洋地黄甙不良反应。
奎尼丁同用时,可因减少地高辛肾清除率和改变组织分布容积而增高Digoxin血浓度,增加不良反应发生;可增加Digoxin血浓度,从而增加中毒危险性的药物尚有乙胺碘呋酮、维拉帕米、硝苯吡啶。
二、非强心甙类正性肌力药
70年代以来有一系列非强心甙类增加心肌收缩力的药物陆续引入临床应用(表22-4),是近年心力衰竭治疗上一个方面的新进展。其中儿茶酚胺类的多巴胺、多巴酚丁胺、吡丁醇以及双异吡啶类的氨吡酮(Amrinon)、咪利酮(Milrinone)可静脉给药,作为急诊强心治疗。
表22-4强心药的作用机制
| 药 名 | 主 要 作 用 机 制 |
| 1.洋地黄强心甙 | Na-K-ATP酶抑制 |
| 2.儿茶酚胺 | |
| 静脉 去甲肾上腺素 | β[XB]1[/XB]刺激→↑cAMP |
| 肾上腺素 | |
| 多巴胺 | |
| 多巴酚丁胺 | |
| 对羟苯心安(premalterol) | β[XB]1[/XB]兴奋 |
| Butopamine | |
| 口服 吡丁醇 | β[XB]1[/XB]及β[XB]2[/XB]兴奋 |
| 舒喘灵 | |
| H80/63 | |
| 异波帕胺(Ibopamine) | β[XB]1[/XB]兴奋及部分β[XB]1[/XB]抑制 |
| 3.双吡啶类 | |
| 胺吡酮 | ↑Ca[SB]2+[/SB]运动 |
| 咪利酮(Milrinone) | 磷酸二酯酶抑制 |
| Enoximone | |
| 4.黄嘌呤类 | |
| 氨茶碱 | 磷酸二酯酶抑制→cAMP↑ |
| 5.钙 | ↑Ca[SB]2+[/SB]运动 |
| 6.胰高血糖素 | 刺激cAMP |
儿茶酚胺的正性肌力作用是通过兴奋心肌β[XB]1[/XB]肾上腺素能受体。肌浆网的β受体受兴奋,激活腺甙环化酶,后者催化APT为3′、5′-cAMP。然后蛋白激酶受cAMP激活,使多膜系统磷酸化作用增加,结果增加钙离子逆转,更多的钙与收缩蛋白作用而产生正性肌力效应。β[XB]1[/XB]受体兴奋同时也增加窦房释放和房室传导,致心动过速及(或)心律失常可能出现,因而可能限制这类制剂最大正性肌力作用的发挥。各种拟交感胺制剂对各种交感胺受体的作用不同(表22-5)。
(一)多巴胺(Dopamine)
1.药理作用 多巴胺为去甲肾上腺素的直接前体。多巴胺兴奋三种不同的受体:①兴奋多巴胺受体,小剂量(1~5μg/kg·min)即有突出效应,使肾血管、肠系膜及内脏血管扩张,血流增加,尿量增加。②直接兴奋心肌β受体(5~10μg/kg·min),也使去甲肾上腺素释放,产生正性肌力作用,使心排血量持续增加,心率仅轻微增加。③作用于α[XB]1[/XB]受体,引起血管收缩,血压增高,小剂量时此效应不显,当剂量小于10μg/(kg·min)时,增高血压作用明显,可产生心动过速、心律失常;当剂量大于30μg/(kg·min),对周围血管作用似去甲肾上腺素,但不影响肾血流量。
表22-5心脏、血管的交感胺受体和拟交感胺制剂的作用
| 受 体 | 肾 上 腺 素 能 受 体 | 多 巴 胺 受 体 | |||||
| β[XB]1[/XB] | β[XB]2[/XB] | α[XB]1[/XB] | α[XB]2[/XB] | 1 | 2 | ||
| 部位 | 轴突后 | 轴突后 | 轴突后 | 轴突前 | 轴突后 | 轴突前 | 轴突前 |
| 效应 | 心肌兴奋 | 血管扩张 | 血管收缩 | 阻抑去甲肾上腺素释放 | 血管收缩 | 血管扩张 | 阻抑去甲肾上腺素稀放 |
| 多巴胺 | ++ | + | ++ | +++ | |||
| 多巴酚丁胺 | ++ | + | + | + | |||
| 对羟苯心安 | ++ | ○ | ○ | ||||
| 去甲肾上腺素 | ++ | + | ++++ | - | ++ | ||
| 肾上腺素 | +++ | +++ | +++ | + | ++ | ||
| 异丙肾上腺素 | ++++ | ++++ | ○ | ||||
| 间羟胺 | ○ | ++++ | |||||
| 沙丁胺醇 | + | ++++ | ○ | ||||
2.药代学 T[XB]1/2[/XB]为1~2min,故此需持续滴注。受单胺氧化酶作用,75%代谢产物无活性,25%代谢形成去甲肾上腺素。
3.适应证 多巴胺可用于急性心肌梗死的心源性休克,开放式心脏外科手术和术后,创伤,内毒素性败血症,肾功能衰竭,对少尿、周围血管阻力降低或正常的病人更有益。对心源性休克,应在低血容量矫正后使用多巴胺。对已显示或正在发展过度血管收缩的病人宜与酚妥拉明或硝普钠合用,偶也可与异丙肾上腺素合用。
4.禁忌证 嗜铬细胞瘤、甲状腺功能亢进。当考虑期望的益处大于对胎儿的危险性时,多巴胺也可用于妊娠期。
有周围血管病史(如动脉硬化、动脉栓塞、冻创、雷诺病或Büerger病等),应用多巴胺时应密切监测肢体皮肤颜色或温度改变,避免肢体缺血或坏死,有时需减少剂量。
5.剂量和用法 多巴胺20mg加入5%葡萄糖液(或其溶液)100~200ml,静滴,自小剂量开始。根据体重及治疗反应调整浓度和速率。小剂量(2~5μg/kg·min)使尿量增加,心排血量不变或轻度增加;中等量(6~10μg/kg·min),增加心排血量,尿量维持,开始使心率、血压增加;较大剂量(11~20μg/kg·min)使心排血量增加明显,心率和血压增加,肺毛细血管压增加,可致心律失常,应用多巴胺过程需适当补充血容量,因尿量较大;停药时应逐步减少剂量再停用,防止低血压发生。
6.不良反应 最常见有心悸(由于期前收缩或心动过速)、恶心、呕吐、心绞痛、头痛、低血压、血管收缩表现(肢冷、脉压减小),其他如心动过缓、QRS增宽、血压增高、氮质血症等。
7.药物相互作用 与单胺氧化酶抑制剂合用时,多巴胺剂量应减少(前药可加强多巴胺作用),初始剂量可为通常剂量的1/10。
多巴胺增强利尿药作用,因它扩张肾血管,增加肾血流量和肾小球滤过液以及钠排泄。
多巴胺拮抗吗啡的镇痛作用。
多巴胺可与异丙肾上腺素、酚妥拉明或硝普钠合用。对利尿药或多巴胺反应差的病人,多巴胺与利尿药如速尿合用,可产生较好的利尿效果。
如多巴胺不能维持血压,可与间羟胺合用。
接受多巴胺的病人静脉给予苯妥英钠,可发生低血压和心动过缓,宜避免合用。
[b](二)多巴酚丁胺(Dobutamine,Dobutrex[SB]®[/SB])[/b]
1.药理作用 为一种合成的儿茶酚胺。①直接增强心肌收缩性(β[XB]1[/XB]受体兴奋),作用强度相等于异丙肾上腺素,但变时性效应和增强周围阻力效应明显较小,为其优点。剂量小于7.5μg/(kg·min),心排血量增加,不增快心率和血压。②可降低肺毛细血管压,不像多巴胺甚至可增高肺毛细血管楔压。③剂量大于7.5μg/(min·kg)可微弱兴奋α受体,偶有轻度血管收缩,通常周围血管阻力降低。最适用于正常血压病人,继发于心排血量降低的中度低血压,因其可增加心排血量。显著低血压患者宜与加压药合用,不宜单用。多巴酚丁胺与多巴胺对心力衰竭患者血流动力学效应的异同见表22-6。
表22-6 多巴酚丁胺与多巴胺对心力衰竭患者的血液动力学效应的比较
| 心排血量 | 动脉压 | 周围循环阻力 | 左室充盈压 | 心率 | 每搏排血量 | 心搏作功指数 |
| 多巴酚丁胺↑↑ | — | —↓ | — | —↑ | ↑↑ | ↑↑ |
| 多巴胺↑↑ | ↑ | —↑ | —↑ | ↑ | ↑↑ | ↑↑ |
—表示不变, ↓↓或↑↑表示明显减少或增加。
2.药代学 1~2min内生效,达高峰作用一般需10min;血浆T[XB]1/2[/XB]为2min。主要代谢途径为儿茶酚甲基化和结合。人尿主要排泄产物为多巴酚丁胺的3—0—甲基多巴胺的葡萄糖醛酸盐,后者无活性。
3.适应证 心肌收缩力减弱所致成人心力衰竭,作为正性肌力支持治疗,可静脉短时给予(24、48至72h)。急性心肌梗死病例应小心应用。
4.禁忌证 肥厚性心肌病;心房颤动应先用洋地黄控制心室率;原有高血压,可致血压严重增高;妊娠,其对胎儿作用尚未明确。
5.用法、剂量 对大多数病人,多巴酚丁胺2.5~10μg/(kg·min),可获满意反应,对一些病人0.5μg/(kg·min)有效,另一些需40μg/(kg·min)。最好用输液泵控制给药。
每小瓶含多巴酚丁胺250mg或100mg,可加入5%葡萄糖液、生理盐水或N/6乳酸钠溶液中,溶液呈粉红色,随时间变深,不说明药效显著丢失。可按表22-7稀释。
表22-7多巴酚丁胺配制参考表
| 最后浓度 | 100mg | 250mg |
| 加入溶液量 | 加入溶液量 | |
| 1000μg/ml | 100ml | 250ml |
| 500μg/ml | 200ml | 500ml |
| 250μg/ml | 400ml | 1000ml |
| 200μg/ml | 500ml |
用药期间应持续监测心电图、血压,最好能监测肺动脉楔压和心排血量。用药前应用适当扩张血容量以矫正低血容量。
6.不良反应 常见恶心、头痛、心绞痛及非特异性胸痛,可引起心率增快,血压(尤其收缩压)增高。可引起异位心律,如房早、室早,偶见室性心动过速。过敏反应如皮肤红斑、发热、嗜酸粒细胞增多,偶有支气管痉挛。
7.药物交互作用 不应与β受体阻滞剂如心得安合用,因后者可干扰其作用。多巴酚丁胺可与洋地黄、亚硝酸类、利尿药、利多卡因等合用。
三、利尿药
急诊选用的利尿药常要求作用迅速、强效,在各类利尿药中以髓襻利尿药如利尿酸、呋喃苯氨酸、布美他尼等符合上述要求。
(一)呋喃苯胺酸(速尿,Furosemide,Lasix)
1.药理作用 ①利尿迅速、效强,但相对短暂。主要作用于肾小管髓襻升支髓质部,抑制Cl[SB]-[/SB]主动重吸收,Na[SB]+[/SB]重吸收也随之减少;排Na[SB]+[/SB]、排Cl[SB]-[/SB],也排K[SB]+[/SB],易致低血钾。②其他作用,速尿可刺激肾素分泌,扩张肾皮质血管,增加肾血流量(肾功能不全也适用)。还可能扩张其他器官血管床,静脉给药可改善肺充血,降低左心室充盈压,急性血流动力学效应出现在利尿之前。
2.药动学 肌注速尿30min达高峰,口服则为1h。作用持续4~6h。口服可吸收但不完全,蛋白结合率95%~99%,可诱过胎盘和自乳汁分泌。经肾排泄,大部分为原型,10%代谢,与葡萄糖醛酸结合,也经胆汁排泄。T[XB]1/2[/XB]为30~70min,心衰、肾衰时延长。
3.治疗应用 适用于急性肺水肿或充血性心力衰竭、高血压急症、肾病综合征或肾硬变性水肿。对其他利尿药无效病例可能有效。
急诊常肌注或缓慢静注(1~2min),初剂20~40mg,至少等1~1.5h后,视情况再给第2剂。大剂量用于急性或慢性肾功能衰竭,有时用量达1000mg/d。10天以上的长期用药,利尿效应减弱。宜间歇疗法,给药1~3天,停用2~4天。每支20mg(2ml),片剂20mg。
4.不良反应 ①液体和电解质失衡:常见如低钠血症、低钾血症、低镁血症、低血容量可伴低血压。可出现电解质紊乱的一系列症状,如食欲不振、恶心、呕吐、疲劳、乏力、口渴、头晕、肌痉挛等。②其他代谢改变:高尿酸血症、高血糖症、尿糖阳性等。③不常见的不良反应,如胰腺炎、高渗性昏迷、光感性皮炎、皮肤红斑、骨髓抑制、颗粒细胞减少、血小板减少、暂时性耳聋、听力减退(血药浓度大于50μg/ml)。
5.药物相互作用 速尿抑制肾脏排泄庆大霉素、头孢菌素和地高辛,当与前两者合用时,可增加其肾和耳毒性,在肾功能衰弱时,此相互作用更易发生。速尿产生的低血钾增加洋地黄中毒的发生率和危险。
速尿可干扰水杨酸盐排泄,两者合用时,有时低剂量即可出现水杨酸中毒。
妊娠初3个月最好避免用速尿,前列腺肥大和排尿困难病例宜慎用。
速尿不应与强酸性溶液混合。
(二)利尿酸(Ethacrynicacid,Edecrin)
1.药理作用 似速尿,为襻利尿药,作用强。
2.药代学 利尿作用,静注10min左右出现,持续2h;口服30min后生效,2h达高峰。作用持续6~8h。95%血浆蛋白结合。分布于肝浓度较高,主要由近曲小管分泌和经胆汁排泄。在体内无蓄积作用。
3.治疗应用 治疗各种水肿。每支含利尿酸钠25mg,急性肺水肿时可以25~50mg利尿酸钠溶于20~40ml5%葡萄糖液或生理盐水,缓慢静注,或静点。
必需注意补钾。
4.不良反应 似速尿。如电解质紊乱,胃肠道症状常见;厌食、恶心、呕吐、腹泻、可致胃肠道出血;晕眩、暂时性或永久性耳聋。耳聋可出现在静注100mg后1h内,应避免与氨基糖甙类抗生素合用。少数病人可致肝细胞损害、粒细胞减少、皮疹等。
(三)布美他尼(丁尿胺,Bumetanide,Bumex)
1.药理作用 为较新的速尿类衍生物。作用机制似速尿。利尿效应较速尿强20~40倍,因而剂量较小。失钾作用较速尿轻。有扩张肾血管作用。
2.药代学 可静脉注射,数分钟后开始利尿,30~60min达高峰,作用持续2~4h。T[XB]1/2[/XB]为1~1(1/2)h。口服吸收迅速,30min显效,1~4h达高峰,持续4~6h。蛋白结合率大于95%。分布容积0.2~0.3L/kg。部分代谢,部分以原型自尿排出。
3.治疗应用 用于各种顽固性水肿和肺水肿、急性左心衰竭可静注或肌注0.5~1mg(每支2ml含0.5mg),必要时30min再给1次。口服1~10mg/d,分次给(每片1mg)。
4.不良反应 同速尿,糖尿发生率低。不宜加入酸性溶液中静点,以避免发生沉淀。
四、抗心律失常药
目前临床应用的抗心律失常药分类是根据药物主要电生理作用而区分的(表22-8)。
第Ⅰ类钠通道阻滞剂,也称膜抑制剂,有局麻作用,对心肌细胞膜电位。相有直接抑制作用,此外有抗胆碱能作用,通过植物神经间接影响传导系统,减慢传导;对动作电位时间,4相除极电位坡度也有不同程度影响,根据其影响程度又可分为Ⅰ[XB]A[/XB]、Ⅰ[XB]B[/XB]、Ⅰ[XB]C[/XB]三亚类型。
表22-8 抗心律失常药分类—依据其作用机制
| 分类 | Ⅰ类 | Ⅱ类 | Ⅲ类 | Ⅳ类 | 其他 | ||
| A | B | C | |||||
| 主要作用 | 钠通道阻滞剂 | 交感阻滞 | 延长除极 | 钙通道阻滞 | |||
| 抑制O相 | 抑制O相 | 抑制O相 | |||||
| 中度 | 轻 度 | 显 著 | |||||
| 减缓传导 | 减缓传导 | 减缓传导 | |||||
| ++ | 0~+ | ++++ | |||||
| 延长除极 | 缩短除极 | 对除极很少作用 | |||||
| 药物 | 奎尼丁 | 利多卡因 | 英卡胺 | β阻滞剂 | 胺碘酮 | 维拉帕米 | 洋地黄 |
| 普鲁卡因胺 | 苯妥英钠 | 氟卡胺 | 普萘洛尔 | 溴苄胺 | 硫氮[img]/assets/zyimg/shu/jizhenyixue/jizhenyixue054.jpg[alt][/alt][/img] 酮 | 钾盐 | |
| 双异丙吡胺 | 氨酰甲苯胺 | 氯卡胺 | 氨酰心安等 | 异丙肾上腺素 | |||
| 美心律 | 茚满丙胺 | 烯苯胺咪 | |||||
| 乙吗噻嗪 | 丙胺苯丙酮 | ||||||
| 缓脉灵氯乙酯 | |||||||
急诊常用的有利多卡因、奎
■[此处缺少一些内容]■
结影响小。②治疗剂量的利多卡因极少引起症状性或临床主要的血流动力学异常,可安全用于已洋地黄化的心衰患者。③利多卡因可影响中枢神经系统,小剂量具有镇静、中枢镇痛及抗惊厥作用,过大剂量则引起惊厥及呼吸停止。
2.药代学 利多卡因口服虽可吸收,但因其高度的肝清除率,故只宜静脉或肌注给药。静脉入壶,3min内即达峰浓度,持续10~20min。呈二室模型分布。消除半减期1~2h。利多卡因抗心律失常有效治疗血浓度为1.5~5μg/ml(即6.5~21μmol/L),浓度为3~5μg/ml时治疗作用与致毒作用交叉,大于6μg/ml(25~26μmol/L)常出现中枢神经中毒症状。心力衰竭、活动性肝病时利多卡因清除率降低,半减期延长,易出现中毒症状。利多卡因静滴24h,半减期也延长,可达4h,故宜减量使用。
3.适应证 ①各种室性早搏,特别是频发(5次/min以上),成联律或多源性,发生于T波顶峰;各种急诊情况,如急性心肌梗死、心导管检查或心外科手术时,疗效佳;②室性心动过速,效佳;③洋地黄中毒或电复律后的室性快速性心律失常;④室颤引起的心脏停止,效佳。
4.禁忌证 ①对胺类麻醉药过敏者;②高度窦房、房室或心室内阻滞;③窦性心动过缓伴室性(或房室交界性)逸搏,除非预先用异丙肾上腺素使心率增快,否则利多卡因可增加逸搏频率,或出现更严重的室性心律失常;④房颤伴差异性传导(QRS畸形),误给利多卡因可增加房室传导而增快心室率。
5.剂量用法 针刺,每支0.2g(10ml)或0.4g(20ml)。可由小壶入,静滴剂量为先50~100mg,小壶入(不稀释),或每15min50mg,必要时重复1~2次;同时静滴,速率1~3mg/min(即100~300mg利多卡因加入5%葡萄糖液100ml中),每分钟1ml或用恒速输液泵调节。
6.不良反应 与剂量有关,通常发生于剂量在200~300mg/h以上时。局部可发生血栓静脉炎。
神经系统可有头晕,激动或欣快,倦睡,耳鸣或听力减退,视物模糊或复视,呼吸、说话或吞咽困难,热、冷或发麻感觉,呕吐,肌肉震颤,局部或全身抽搐或发生惊厥,神志不清,呼吸抑制甚至呼吸停止。
心血管系统通常不受影响,但过量时可产生低血压、休克、心动过缓、完全性房室阻滞、窦房阻滞或心脏停顿。
如发生严重反应,应即中止给药;如有搐搦,可用超短作用的巴比妥盐,如硫贲妥钠0.1~0.2g或安定10mg静注。
7.药物相互作用 利多卡因与普鲁卡因酰胺间或利多卡因与奎尼丁间的交叉敏感罕见,但可发生。利多卡因与普鲁卡因同用,可增加中枢神经敏感性,产生烦躁不安、幻视或其他症状。
心得安可增加利多卡因毒性。甲氰咪胍也可增加利多卡因毒性。
1.药理作用 属Ⅰ[XB]A[/XB]类抗心律失常药。①也直接作用传导系统,减缓AV传导,可延长P-Q间期,延长心室肌动作电位时间,可增宽QRS及QT间期,QRS较对照值增宽25%至50%为毒性症状。②有抗胆碱能作用,可加快心室率,特别于房颤或房扑时。③有β受体阻滞作用,在严重心脏病,快速性心律失常伴低血压,奎尼丁对心肌抑制作用。④α受体阻滞作用,使周围血管对α肾上腺素能兴奋剂不起反应,故奎尼丁引起的低血压严重。
2.药代学 口服吸收良好。口服单剂,达峰时间1~3h,作用持续6~8h,T[XB]1/2[/XB]为6h,老年人延长(9.7h左右)。80%与血浆蛋白结合,经肝氧化途径消除。10%~20%以不变形式经尿排出。充血性心衰或肾功能不全时,奎尼丁尿排泄减慢,血中游离型浓度较高,宜减少剂量。碱性尿延缓奎尼丁排泄,酸性尿加速其排泄。治疗范围的血浆奎尼丁浓度为1.5~5μg/ml(平均3.5μg/ml),宜测峰值和谷值(末次剂量后1h后6~8h取血,如为奎尼丁葡萄糖醛酸盐,峰值在口服后4~8h)。肾功能不全时,奎尼丁血浓度可升高,但并不提示奎尼丁毒性。
3.适应证 ①房性早搏,效佳。②阵发性房性心动过速,效佳。③房颤转复和维持窦律,效佳。转复过程可出现暂时性房扑(如用药过程中房扑持续,应停奎尼丁)。为预防其阻滞迷走神经作用而增快心室率,宜先给速效洋地黄。④预激综合征合并阵发性房颤,效佳。⑤房扑转复(洋地黄化后),效可。⑥交界性早搏,效好。⑦交界性阵发性心动过速,效好。⑧室性早搏,室性心动过速,效好至效佳。⑨洋地黄所致室性心动过速,效可。
4.禁忌证 ①室颤,奎尼丁减低室颤波幅度,增加电复律电能;②Ⅱ度或完全性传导阻滞;③对奎尼丁或奎宁类药物严重不良反应或毒性史。
5.剂量、用法 硫酸奎尼丁,每片0.2g,主要口服。为复律目的。第一天0.2g,每2h一次,共5次。如无效也无毒性反应,第2天增至0.3g,每2h一次,连续5次,每日总量一般不超过2g。每次给药前应测血压与心电图。恢复正常心律后,即给维持量。一般为有效量减去0.2g,2~3次/d。奎尼丁有效剂量与中毒剂量之间距离甚狭窄。不宜静脉用奎尼丁。
6.不良反应 最常见不良反应为恶心、呕吐、腹泻、头晕或头痛、耳鸣、视力障碍(金鸡纳反应)。如不严重一般可不停药,其发生与血药浓度无相关。心血管系统可有心动过缓、心力衰弱、低血压、休克、完全性房室传导阻滞、心室停搏、扭转型室性心动过速、室颤等。晕厥为严重不良反应,由于尖端扭转型室性心动过速,可致猝死。奎尼丁晕厥甚至可发生于治疗血浓度范围内。过敏反应如血管神经性水肿、特异质反应、潮红与呼吸困难、丘疹等。
7.药物相互作用 奎尼丁抑制肾小管对地高辛的排泄,使地高辛稳态血浓度倍增。两者合用时,地高辛量应减半。苯巴比妥、苯妥英、利福平等增快奎尼丁的代谢清除,合用时奎尼丁量需增大。奎尼丁不宜与延长QT间期的药物(如普鲁卡因胺、胺碘酮、双异丙吡胺等)合用。利尿剂等引起的低血钾可减弱Ⅰ型抗心律失常药的效应,应用奎尼丁时应先纠正低血钾。
(三)普鲁卡因胺(普鲁卡因酰胺,Procainamide,Pronestyl,简称PA)
1.药理作用 属Ⅰ[XB]A[/XB]型抗心律失常药,似奎尼丁。降低心肌自律性,减慢房室传导速度,延长动作电位时间和不应期,α肾上腺素受体拮抗作用。可发生低血压,可减弱心肌收缩力,增高左心室舒张期终末压,也降低心排血量和增高肺动脉压。
2.药代学 口服后几乎完全吸收。生物利用度为剂量的75%~95%。口服后1h,肌注后30min,血浓度达高峰。静脉单剂,符合开放式二室模型。平均T[XB]1/2[/XB]α为5min;T[XB]1/2[/XB]β为3h(2.2~5.0h),晚期肾功能不全显著延长。PA约50%在肝代谢,经乙酰化而形成主要代谢产物。肝疾病对PA代谢减弱,应减小PA剂量。充血性心衰病人也应相应减少负荷与维持剂量。治疗血浓度为4~8μg/ml(占85%病例),另10%有效病例血浓度为8~12μg/ml。
3.适应证 急、慢性房性或室性心律失常,特别对室性心律失常更有效,如室性早搏、室性心动过速效佳;用于洋地黄所致室上速不伴AV阻滞、室速效好。
4.禁忌证 Ⅱ度或Ⅲ度AV阻滞、重症肌无力、过敏反应者(与普鲁卡因可能产生交叉过敏反应)。
5.剂量、用法 紧急复律先用静注或静滴。静注为3~5min内静注100mg,每隔5~10min重复一次,直至有效,或总量不超过10~15mg/kg;也可静滴;0.5~1g溶于5%~10%葡萄糖液100~200ml,开始10~30min速度可适当较快,于1h内滴完,无效者,1h后可再给1次。总量不超过2g/d。
6.不良反应 ①恶心、呕吐、厌食,口服时常见。②心脏方面有低血压,甚至休克,尤其静脉用药时;可发生室内传导阻滞、QT间期延长、室性心动过速、心室颤动、停搏、心衰等。③特异体质病人可发生过敏反应,可见皮疹、发热、肌肉痛、粒细胞减少、血管神经性水肿等。毒性处理为首先停药。严重低血压有时需用升压药。明显QRS增宽伴心率减慢,可静脉给予碳酸氢钠。
7.药物相互作用 可与心得安合用,两药剂量宜减少。可与洋地黄合用。不宜与延长QT间期的其他药物如奎尼丁、胺碘达隆等合用。与利尿药、降压药合用时可加重低血压作用。PA可增强神经肌肉阻滞剂作用,合用时可产生严重呼吸抑制。
(四)双异丙吡胺(Disopyramide,Rythmodan,Norpace)
1.药理作用 ①似奎尼丁,具有Ⅰ类直接抑制细胞膜作用(属Ⅰa类),缩短、减低V[XB]max[/XB],抑制4相除极坡度,延长动作电位时间。②有较强的抗胆碱能作用。③有负性肌力作用,减弱左心室射血分数和心排血量,心储备功能降低时可诱发心力衰竭,尤易发生于静脉用负荷量时。
2.药代学 磷酸盐口服吸收迅速、完全(90%),峰浓度出现在2~3h后,血浆蛋白结合率50%~80%;表观分布容积0.5L/kg;约50%以原型自尿排出,部分经N-脱羟基作用代谢,代谢物具有突出的抗胆碱能作用。T[XB]1/2[/XB]6~10h,肾功能不全时延长。通常有效治疗血浓度为3~5μg/ml,其蛋白结合率为浓度依赖性。游离血浓度对不良反应呈线性关系,应用时应监测游离型血浓度。
3.适应证 可用于室性和室上性心律失常;对室性早搏、持续性和非持续性室性心动过速有效。长期口服,有效率50%以上。对自然发生的室速,静注后75%~95%终止发作。对20%~66%程序刺激诱发的室速有预防作用。对室上速效果较差。
4.禁忌证 心源性休克;原有Ⅱ度或Ⅲ度房室传导阻滞(未安装起搏器者);已知对本药过敏者;病窦综合征。前列腺肥大、轻度心力衰竭慎用。
5.剂量、用法 50~100mg稀释后静注,5~10min内缓慢注完,必要时45~60min后可再给1次;静滴,100~200mg以5%葡萄糖液500ml稀释,速度不宜太快,一般0.5~1mg/kg·h(即20~30mg/h),每支50mg(5ml)。口服100~150mg,每日3次。每个胶囊为100mg或150mg。缓释胶囊(Norpace C[XB]R[/XB])为100mg或150mg,每12h一次。
6.不良反应 ①口干、视力模糊、排尿困难常见(抗胆碱作用所致)。②主要副作用在心脏方面,可产生严重心力衰竭或心源性休克,慢性口服者也可发生。偶可引起多形性室性心动过速、室颤。发作前QRS增宽,QT间期延长。开始用药即应EKG监测,一旦发现QRS增宽即应停药。③胃肠道可发生恶心、呕吐、腹泻,可有肝功能损害,出现黄疸。④神经系统有头晕、乏力、倦怠等急性精神症状。⑤其他如低血糖。
7.药物相互作用 与Ⅰ类抗心律失常药合用时应防其相加作用,发现QRS增宽,应停药。与β受体阻滞剂心得安等合用,可加重负性肌力作用,诱发心衰。与地高辛合用可改善本品负性肌力作用。与苯妥英钠合用,可加速本品代谢,应增大剂量。
(五)茚满丙二胺(安搏律定,Aprindine,Aprinidine)
1.药理作用 属Ⅰ[XB]C[/XB]类抗心律失常药。对窦房结影响较小,减慢心房及房室传导,延长A-H和A-V间期及其不应期。缩短浦肯野纤维动作电位时间,使有效不应期缩短,有阻断房室旁路传导作用,能延长旁路不应期。很少影响植物神经系统。静脉用药使心
■[此处缺少一些内容]■
50mg,每日3次;以后每日50mg,每日2次,或每次25mg,每日2~3次维持。也有报道首次100mg,以迅速提高血浓度,以后再按一般剂量服用。每片25mg或50mg。静滴时首次100~200mg,用5%~10%葡萄糖液100~200ml稀释,滴速为2.5(2~5)mg/min,必要时30min及6h可分别再给100mg,24h总量不超过300mg。次日改为口服。急诊病例可在心电图监测下滴速可增加为10mg/min。密切观察QRS,若增宽15%应减慢滴速;如需静滴维持,也须逐日减半剂量,50~100mg/d为维持剂量。
6.不良反应 有效治疗浓度范围很窄,易发生不良反应。①中枢神经系统反应有眩晕、感觉异常、颤抖、复视、幻觉,严重时可出现癫痫样抽搐,停药3~4天可恢复;②胃肠道反应;③剂量过大或注射速度过快可引起QRS增宽,P-R间期轻度延长,停药15~20min可逐步恢复,严重时可出现室性心动过速或室颤;④也有出现粒细胞减少症(常于初始4~6周出现)和黄疸的报道,应定期复查WBC、肝功能。
(六)丙胺苯丙酮(心律平,Profafenone,Rytmonorn)
1.药理作用 ①属Ⅰ[XB]C[/XB]类,为表面麻醉药。降低细胞O相上升速度(V[XB]max[/XB]),轻度延长动作电位时间和有效不应期。一次静脉给药,A-H、H-V、心房、心室有效不应期、A-V传导时间均延长。QRS波群、P-R间期增宽,但多在正常范围内,超过正常者与剂量过大、本身有器质性心脏病有关。QT间期延长20%左右,说明对整个传导系统均有抑制作用,也延长旁路传导。②有轻度抑制心肌作用,增加舒张末期压,减少搏出量,作用依赖剂量。
2.药代学 口服后吸收良好,2~3h达高峰血浓度。呈二室模型。半减期平均3h,但在体内代谢有强弱之分,T[XB]1/2[/XB]长于10h者为弱代谢型,大部分为强代谢型。治疗血药浓度个体差异大。增加剂量,血浓度呈非线性上升。
3.适应证 对室上性和室性心律失常均有较好疗效。室早有80%以上病例完全抑制,室性心动过速控制率90%以上。对抑返性室上性心动过速效果比房性心动过速为好,至少可减慢心室率。对阵发性房颤、房扑、半数可转复,尤其WPW综合征伴房颤时,可明显减慢心室率,并可预防再发。
4.禁忌证 严重心力衰竭、心源性休克、严重窦缓、病窦综合征,明显电解质失调,严重阻塞性肺病患者,明显低血压禁用。
5.剂量、用法 静注,必要时在ECG监护下进行,70mg(每支70mg/20ml)于3~5min内注入,间隔10~20min可再给一次,最大量不超过350mg。也可按0.5~1mg/kg静注后,改口服。口服首次量300~600mg,以后600~900mg/d,分2~3次服用。片剂每片为50mg或150mg。
6.不良反应 较少。①恶心、呕吐等轻度胃肠道反应,可有味觉障碍;②轻度阻滞β受体作用,运动心率下降,收缩期血压降低,P-R延长(减慢心率作用不及心得安的1/4),于服药后1~2h消失;③有报道出现心功能损害,心力衰竭,个别出现束支和房室传导阻滞,室性心律失常发作较频繁。
7.药物相互作用 与地高辛同服时,本品可使地高辛血浓度增高。
(七)美心律(慢心律,Mexiletine)
1.药理作用 似利多卡因,属I[XB]B[/XB]型抗心律失常药。可口服,抑制心肌快反应及膜反应性,缩短动作电位时间(APD),延长有效不应期(ERP)及ERP/APD比值。对植物神经系统无明显作用。
2.药代学 口服生物利用度高,近90%;2~4h达血浓度高峰;分布容积9L/kg,与血浆蛋白结合率中度(70%),消除T[XB]1/2[/XB]9~12h;有效治疗血浓度0.5~2μg/ml。约10%经尿排泄。
3.适应证 治疗,尤其预防室性快速性心律失常(如急性心肌梗死时,二尖瓣脱垂,QT延长综合征、洋地黄中毒等),对房性快速性心律失常可能无效。
4.禁忌证 中度以上传导阻滞、病窦综合征、严重心动过缓;心力衰竭时慎用。
5.剂量、用法 静注:70~100mg(针剂每支100mg/2ml)加入5%葡萄糖液20ml,缓慢静注(3~5min),如无效,隔10~20min重复一次,以后以1.5~2mg/min,静滴3~4h后,减慢速度0.75~1mg/min,并维持24~48h。口服:片剂每片50mg或100mg。一般每次150mg,每6~8h一次,有效后酌情减量,每次100mg,每日3次。
6.不良反应 中枢神经系统症状有头晕、震颤、复视、嗜眠等;消化道症状有恶心、呕吐;心血管反应有低血压、心动过缓、房室传导阻滞,尤其静脉用药后。
7.药物相互作用 其他局麻性抗心律失常药可加强其效应;可与β受体阻滞剂、地高辛安全合用;麻醉性止痛药可延缓其吸收。
(八)普洛萘尔(心得安,Propranolol,Inderal)
1.药理作用 为一种最常用的β受体阻滞剂。心脏仅有β[XB]1[/XB]肾上腺素能受体,对去甲肾上腺素、肾上腺素(或异丙肾上腺素)起反应,结果产生正性肌力作用(增加收缩强度)和正性变时性效应(增快心率)。心得安于心肌及心脏传导组织β受体部位与儿茶酚胺进行竞争,结果产生负性肌力和负性变时变性效应,也增加肾小管钠重吸收而增加血容量(可致水肿及其他充血性心衰症状)。
心得安具有非特异性抗心律失常作用,属Ⅲ类抗心律失常药。其抗心律失常机制主要通过阻滞对起搏点电位的肾上腺素能激动作用,抑制应激性和传导性,这使交感神经过度刺激,如在急性心肌梗死或洋地黄中毒时产生的心律失常对β受体阻滞剂反应良好;β受体阻滞剂预防心肌缺血,可减少自律性,抑制折返机制。
心得安的负性肌力作用可引起左室舒张末压增高和心排血量降低(可被洋地黄部分抵销);心得安不干扰硝酸甘油类药物的扩血管作用;它可降低血压。
2.药代学 口服吸收良好,肝“首过”代谢显著,生物利用度通常低于40%,峰浓度出现在口服后1~4h,空腹时或进食后吸收更快,作用持续5~6h。心得安治疗血浓度30~100μg/ml,宜于服后1h和1~8h取血测峰值及谷值。如血浓度大于120μg/ml而抗心律失常作用未达到,不应再加剂量而应换药。在人体,心得安85%~95%与血浆蛋白结合。
静脉给药,最大效应发生于10min内,持续约1h?
■[此处缺少一些内容]■
mg,每日3次;嗜铬细胞瘤术前3天服药,每次10~20mg,每日3次;治心绞痛,每次10mg,每日3~4次,逐渐加量至80mg/d或更大。
6.不良反应 主要有①心动过缓;②心力衰竭(诱发或加重);③支气管哮喘(诱发或加重);④乏力、思睡、头痛、失眠、恶心、腹胀、便秘等;⑤周围血管收缩,甚至出现雷诺现象;⑥慢性应用心得安病人如突然停药,可出现心绞痛加重,甚至心肌梗死,宜逐步减量至停用。
7,药物相互作用 心得安等β阻滞剂常与其他抗心律失常药合用;可与洋地黄制剂合用,有助于加强控制房颤、房扑的心室率和减少β阻滞剂的负性肌力作用;与奎尼丁、普鲁卡因胺、利多卡因合用时需减小剂量,以防增强心肌抑制作用。原用利血平一类药,加用心得安可能产生过度交感阻滞;合用心得安可能加强中枢神经抑制药如麻麻醉药、止痛药、巴比妥类、其他镇静药及乙醇等的作用。甲氰咪胍抑制心得安肝代谢,可增高心得安血浓度;维拉帕米等钙拮抗剂与β阻滞剂合用可增强心动过缓,心肌抑制和低血压,特别静脉用药时,不宜合用。心得安可增强胰岛素或口服降糖药效应,甚至产生低血糖。心得安可阻滞甲基多巴的扩血管作用,合用时反可产生高血压(应激时)。与肾上腺素合用时可能产生高血压、宜慎用。
(九)胺碘酮(乙胺碘呋酮,Amiodarone,Cordarone)
1.药理作用 属Ⅲ类抗心律失常药。胺碘酮延长动作电位时间,对整个传导系统,包括旁路均有抑制作用。一次静脉注射(5mg/kg),减慢房室传导,延长A-H间期,增加房室不应期,并轻度延长窦房结复律时间,但不延长H-V间期和心房不应期;长期口服胺碘酮减慢窦率及A-V传导,也减慢希-浦纤维传导,心房、心室及A-V结不应期延长。心电图表现窦缓(不被阿托品拮抗);QT延长。
胺碘酮对血管平滑肌有松弛作用,可降低冠状及周围血管阻力,增加冠状动脉血流。静脉注射过快或量大可明显扩张血管,使血压下降。
本品为含碘化合物,0.2g含碘75mg,可干扰体内碘代谢及甲状腺功能。
2.药代学 吸收不完全,生物利用度20%~40%。口服后达高峰血浓度约4~7h。静注后作用出现快,15s至51min,最大作用为15min~3h,2~6h作用消失。本品为高度亲脂性,体内分布可能为三房室模型,口服4~5天已完成对心肌的分布(浅周边室达相对稳定),即可进行血浓度监测。消除缓慢,T[XB]1/2[/XB]长达15天、3周甚至更长。有效治疗血浓度0.5~2μg/ml(谷值),不良反应与血浓度无直接关系。主要代谢产物为去乙基胺碘酮,其血浓度也随药物剂量而变化。小部分经肝或肾排泄。
3.适应证 对室上性和室性快速性心律失常均有效。阵发性房颤、房扑的预防;房颤转复,未复律者多数心室率明显降低;房室交界性折返性心动过速;伴随WPW的室上速或房颤;对反覆发作室速效果也较好;各种早搏有效;由于室颤、猝死存活者。
4.禁忌证 不宜用于原有重度阻滞或症状性心动过缓(未装起搏器者);甲状腺功能失调者;低心排血量性心力衰竭慎用。
5.用法、剂量 静注,一次量不超过5mg/kg(每支150mg~3ml),以5%葡萄糖液稀释成20ml,缓慢静注5min以上。再次注射间隔15min以上。
口服:国内主张负荷量较小,第1周每次0.2g,每日3次,第2周每次0.2g,每日2次,以后维持量为每日0.2g~0.1g;也可初两周负荷量较大;1.2g/d,分3次给,用2天,以后1g/d×8天,0.6g/d×4天,一日量分次给,再后维持量0.2g/d。每片0.2g。
6.不良反应 长程口服病例不良反应发生率约30%~50%。严重反应有甲状腺功能亢进或低下,需及时停药;肺纤维化;肝功能损害(SGPT增高);心血管方面有低血压(静脉注射过快),心衰可诱发,QT延长(QT[XB]C[/XB]>0.6s需停药)、窦缓、传导阻滞;色素沉着,光敏感;恶心,便秘;视力障碍等。应定期查肝功能、甲状腺功能及X线胸片。
7.药物相互作用 胺碘酮增高地高辛血浓度,合用时应减少地高辛用量。胺碘酮与华法令等口服抗凝药合用时,可延长凝血酶原时间,应减少抗凝药剂量。
(十)维拉帕米(异搏定,戊脉安,Verapamile,Isoptin)
1.药理作用 属Ⅳ类抗心律失常药。特异性抑制慢通道钙(可能还有钠)的内流进入传导或收缩时的心肌细胞和血管平滑肌。窦房结和房室结的电活动取决通过慢通道的钙内流,因而维拉帕米对室上性快速性心律失常非常有效。
类似洋地黄,维拉帕米对伴房颤的WPW综合征病人可缩短不应期和增加心室反应,甚至产生室颤,因而不应给予此类病人。
有扩血管作用,并降低左心室心肌代谢需要,有抗心绞痛作用。
2.药代学 口服吸收良好,近90%,但生物利用度仅10%~22%,说明有明显肝“首过”代谢。呈二房室模型分布。T[XB]1/2[/XB]-α为18~35min,消除T[XB]1/2[/XB]170~440min。70%经尿排泄,16%粪排出。90%与蛋白结合。口服后电生理作用出现于2h,5h达高峰。静注维拉帕米,1~2min内出现效应,10~15min达高峰,维持6h。
3.适应证 静注用于阵发性室上性心动过速,伴或不伴结外途径;房颤或房扑伴快速心室率;由于冠状动脉痉挛引起的室性心率失常。口服用于与洋地黄合用,控制房颤或房扑的快速室率;肥厚性心肌病,如伴左心功能不全、病窦、显著房室结病变者不用。
4.禁忌证 病窦综合征,Ⅱ度或Ⅲ度房室阻滞,心源性休克,低血压(非心动过速所致),与心率无关的心力衰竭,疑洋地黄中毒。避免用于正在服用β阻滞剂、双异丙吡胺、利血平病人,也不用于WPW综合征伴房颤或房扑病人。
5.用法、剂量 静注首剂0.075mg/kg(总量不超过5.0mg),用5%葡萄糖20ml释稀,注射2min以上;如无效或无不良反应,10min后可重复1次上述剂量;仍无效,30min后可给0.15mg/kg一剂。针剂为每支5mg/2ml。口服为每次40mg,每6h一次;2~3天后,需要时渐增量至每次120mg,每6h一次。片剂为40mg/片。
6.不良反应 低血压,有时需用10%葡萄糖酸钙或氯化钙逆转,甚至需用儿茶酚胺;严重心动过缓,重度房室传导阻滞,有时需用异丙肾上腺素、阿托品、起搏器等;心力衰竭,诱发或加重;可有眩晕、恶心、呕吐、便秘等。
7.药物相互作用 维拉帕米可减少地高辛肾清除,增高其血浓度,合用时地高辛应减量;不宜与β受体阻滞剂合用,特别在充血性心肌病、心力衰竭、新鲜心肌梗死时,以防加重心肌抑制。应用维拉帕米前48h和以后24h内不宜与双异丙吡胺合用。
(十一)阿托品(Atropine)
1.药理作用 为阻断M-胆碱受体的抗胆碱药。药理作用多方面。能解除肠道、膀胱及血管平滑肌痉挛和刺激有关的括约肌;抑制汗腺、支气管及其腺体分泌;解除迷走神经对心脏的抑制,增快心率;散大瞳孔,使眼压升高;兴奋呼吸中枢。
2.药代学 口服吸收良好,但经肝“首过”代谢。心脏效应持续2h,其他全身效应可持续24h。
3.适应证 在心脏方面解除迷走神经张力所致窦缓,药物所致心动过缓(如洋地黄、吗啡等);治疗锑剂引起的阿-斯综合征;治疗有机磷中毒;各种内脏绞痛等。
4.禁忌证 前列腺肥大、幽门梗阻、青光眼。
5.用法、剂量 针剂每支为0.5mg或1mg或2mg(以上心脏病常用);尚有5mg及10mg(治疗有机磷中毒用)。皮下或肌注每次0.5mg;静注每次1~2mg,5%葡萄糖10~20ml稀释。口服:片剂为每片0.3mg,每次0.3~0.6mg,每6~8h一次。
6.不良反应 口干,眩晕、皮肤潮红、瞳孔扩大、心率增快、兴奋、谵语、惊厥等。原有前列腺肥大时可引起尿潴留。
(十二)三磷酸腺苷(腺三磷,Adenosinetriphosphate,ATP)
1.药理作用 ATP为一种重要的中间代谢物,参与多种重要生化途径,调节一系列生理过程,包括冠状动脉及体循环血管张力、血小板功能、脂肪组织分解等。
使A-H间期延长,延缓或阻滞房室结的前向传导,阻断折返环路以终止心动过速;房内传导时间、希浦系传导时间等无变化;此外还有较强的拟迷走神经作用,复律后可同时出现窦性停搏及(或)窦性心动过缓。
2.药代学 血浆清除迅速,约30s。作用快,药效消失也快。
3.治疗应用 用于室上性心动过速(维拉帕米无效时可试)。病窦综合征、严重窦缓禁忌;老年人慎用;有过敏史(本品为生物制品)不用。
针剂每支20mg-2ml或粉剂20mg,附磷酸缓冲液2ml。首剂20mg,用葡萄糖液稀释成5ml,5~20s静注,无效者,5min后可再入30mg,单剂量不超过40mg。应用心电图蓝测,观察血压变化和病人反应。
4.不良反应 本品有报道对室上速复律率较高(88.5%),但不良反应发生率也高(90.4%)。可有轻重不等全身不适、胸闷、头晕、头胀、四肢麻木、腹痛、恶心、面部潮红;一般1~2min后可自行缓解;可出现窦停搏、窦缓、Ⅰ~Ⅱ度房室传导阻滞、室性早搏、短阵室性心动过速、房早等;一般对血压影响不大。
(十三)苯妥英钠(Phenytoin,Dilantin,DPH)
1.药理作用 属Ⅰ[XB]B[/XB]类抗心律失常药。在洋地黄中毒时可使房室传导时间和房室结不应期缩短,减少自律性,减小浦肯野纤维4相坡度。皮外,苯妥英钠可能降低心排血量,增加左室舒张末压,可能引起血压下降。抗癫痫,对大脑皮质运动区有高度选择性抑制作用;抗周围神经痛。
2.药代学 口服吸收慢,个体差异大,高峰血浓度出现在2~8h。充分药物作用出现在口服7~10天后。停药后作用仍可持续1周左右。静脉注射作用发生于5~20min。治疗血浓度10~20μg/ml,高于16μg/ml可能中毒。可于静脉负荷量后2~4h,取血测浓度以明确治疗浓度是否已达到。口服药物,1周后开始可测下次剂量前的谷浓度。蛋白结合率高,约90%。5%以原形自尿排出;主要在肝代谢,代谢慢,苯环被肝微粒体羟化,再与葡萄糖醛酸结合,经胆汁、尿排出。其清除取决于肝代谢能力,T[XB]1/2[/XB]在成人一般为24h(20~30h),肝病时可延长。
3.适应证 本品为一种二线抗心律失常药,很少单独应用。目前主要用于洋地黄毒性反应引起的异位心律,如房性心动过速、房颤、房扑、室早、室性心动过速等。
4.禁忌证 伴完全性传导阻滞的室上性心动过速(非洋地黄所致);肝、肾功能衰竭。血浆蛋白低患者慎用,应适当减量。
5.用法、剂量 粉针剂每支0.1g或0.25g。静注每次100mg(注射用水5~10ml稀释),3~5min缓慢注射,每隔5~10min可重复,共3~4次。为维持治疗可改口服,每次0.1g,每日3~4次。
6.不良反应 静脉注射过快可引起低血压、休克、呼吸抑制、心动过缓、传导阻滞、心室颤动、心停搏,甚至猝死。注射速度每分钟不超过50mg,以防其发生。中枢神经反应有头晕、头痛、眼球震颤、运动失调、嗜睡、语言障碍。长期服用可发生恶心、呕吐、厌食、牙龈增生、皮疹、白细胞减少、全血减少或巨细胞性贫血(叶酸缺乏)、紫癜等。
7.药物引互作用 可诱导或抑制肝混合性功能氧化酶的药物均可改变DPH代谢。
与华法令等双香豆素口服抗凝药合用,彼此的血浓度均增高(服DPH,且需口服抗凝剂时,最好换苯茚二酮);阿司匹林、丙戊酸钠等取代血浆蛋白结合,可一过性增加DPH作用;异烟肼、安定药、三环类抑制剂等抑制肝代谢,也增加DPH血浓度,加强其作用,DPH可加强心得安作用。
(十四)异丙肾上腺素(喘息定,异丙肾,Isoproterenol,Isoprenaline,Isuprel)
1.药理作用 为一种强的合成拟交感胺的非选择性β受体激动剂(表22-5);增快心率及心肌收缩性;扩张肾及肠系膜血管,此外,也降低周围血管阻力;增加心排血量,轻度增加收缩压,但降低平均动脉压;对心脏组织的兴奋性和传导性一般增加;增加心肌氧耗量。松弛平滑肌,使变态反应性刺激后的组胺释放减少;松弛妊娠子宫。在人体的主要代谢作用为使游离脂酸稀放。临床作为支管扩张剂、心脏刺激剂以增加房室传导和抗休克药用。
2.药代学 口服吸收完全,但有明显的“首过”代谢。在小肠经硫化作用。T[XB]1/2[/XB]为2.5h。静脉给药的代谢情况不同于口服或吸入。静脉注射后,66%以原形排出,其余游离或结合为3-间-甲基代谢物。口服后,排出的原形占6%~10%;3%~11%以主要结合型3-间-甲基异丙肾排出,其余80%为与硫酸结合的异丙肾形式。3-间-甲基异丙肾为一种非常弱的β肾上腺受体拮抗剂。舌下给药,于15~30min开始作用,持续45min至2h。
3.适应证 抗休克,特别当感染性休克伴有周围血管收缩时;Ⅲ度房室传导阻滞,包括在急性心肌梗死时;治疗哮喘。
4.禁忌证 急性心肌梗死心源性休克时因本品有增加心肌氧耗量作用,一般主张不用。伴血容量不足情况应先扩张血容量,再用异丙肾。
5.剂量、用法 抗休克,在初步扩张血容量后,用0.2~0.4mg异丙肾加入200ml溶液,静滴(每支1mg/2ml)。根据心率反应调整速率。高度房室传导阻滞,心率低于40次/min,0.5~1mg加入5%葡萄糖液200~300ml,静滴。轻者可10mg舌下含服,每日3次。每片10mg。
6.不良反应 心动过速;心律失常(室早、室性心动过速);心悸、头痛、头晕、面部潮红、肌肉震颤、恐惧等。为控制毒性必要时可用β受体阻滞剂。
7.药物相互作用 与其他拟交感胺药物(如肾上腺素等)、氨茶碱等合用可能增加其毒性。
(十五)钾盐
1.药理作用 体内钾大部分集中细胞内,是细胞内主要阳离子,参与细胞内渗透压和酸碱平衡调节、糖及蛋白质合成和能量代谢、神经冲动传导以及心肌和骨骼肌收缩等。心肌细胞内、外钾浓度对心肌的自律性、传导性和兴奋性都有影响。低血钾增加浦肯野纤维自律性,增加静止膜电位和复极速率,使机体对洋地黄敏感性也增加,可诱发多种心律失常;高血钾抑制心肌自律性、传导性和兴奋性。
2.药代学 正常成人自食物摄取钾,每日约2~4g,大部分自肠吸收。正常血钾浓度3.5~5mmol/L。钾离子很快进入细胞内,细胞外液中过量的钾离子迅速经肾由尿排出。
3.适应证 洋地黄中毒引起的阵发性心动过速,如房性心动过速伴房室传导阻滞、交界性心动过速和频发室性早搏;防治低血钾,补充电解质以维持平衡。
4.禁忌证 无尿、高血钾忌用;肾功能严重减退尿少时慎用;除洋地黄中毒外,传导阻滞者慎用。
5.用法、剂量 急诊常用的钾盐制剂有二:氯化钾或门冬氨酸钾镁。氯化钾常用10%氯化钾溶液,每支1g/10ml,含钾20mmol。静滴一般每次用10%~15%溶液10ml,加入5%~10%葡萄糖液500ml稀释,慢滴,溶液浓度一般不超过0.2%~0.4%;为治疗心律失常必要时浓度可加至0.6%~0.7%,即10%氯化钾10ml(或15%7ml)加入葡萄糖溶液200~300ml,在心电图监护下缓慢静滴,多数病例滴注1~2g可见效。滴注过程如心房率开始减慢,预示有效剂量已接近。心律失常控制后改口服制剂。
门冬氨酸钾镁注射液(Potassium magnesium aspartateinjection)为10%溶液,每支10ml含钾2.9mmol,还含镁1.75mmol。门冬氨酸参与柠檬酸循环和鸟氨酸循环,对细胞亲和力强,可作为钾离子载体而使钾离子重返细胞内,相当氯化钾1/4量的门冬氨酸钾,即可充分起治疗作用。10~20ml,加入5%或10%葡萄糖液250~500ml缓慢静滴。
6.不良反应 静滴过量时可出现疲乏,肌张力减低,反射消失,周围循环衰竭,心率减慢甚至心脏停搏。溶液浓度过大可引起局部剧痛、静脉炎。
(十六)镁盐
1.药理作用 镁和钾一样同是细胞内最主要的阳离子。人体镁47%分布细胞内,仅0.4%分布于血浆。在各种与能量有关的活动中,镁是ATP的一种辅因子,也是细胞膜Na[SB]+[/SB]-K[SB]+[/SB]-ATP酶的辅因子。低血钾时此酶活性降低,除非也补充镁,钾返回细胞内也受到限制。硫酸镁注射后可抑制中枢神经系统,松弛骨骼肌,有解痉、镇静、降颅内压作用,可用于惊厥、子痫、尿毒症、破伤风及高血压脑病等。
失钾利尿药过分应用,吸收不良综合征,慢性酒精中毒或其他产生低血钾条件,可致镁缺乏,一般为慢性和发展缓慢。
血镁急剧下降,临床可见于作为应激后,游离脂酸增高的结果,如撤停酒精、急性心肌梗死、心脏搭桥外科手术、暴露于极端寒冷中,应用肾上腺及氨茶碱的后果。洋地黄中毒也伴高发生率的低镁血症,常伴有严重心律失常,需补充镁盐以治疗。
2.药代学 正常血清镁浓度0.75~1.2mmol/L(或1.8~2.9mg/dl)。但即使血镁正常,有时镁盐治疗也有效。硫酸镁虽口服吸收,但治疗心律失常主要用静滴或肌注。
3.治疗应用 用与镁缺乏有关的快速性心律失常、阵发性房性心动过速或室性心动过速、洋地黄产生的心律失常、尖端扭转型室性心动过速。禁忌证为有致高血镁条件者如肾功能不全,心脏传导阻滞。洋地黄化病人小心应用。
硫酸镁常用为每支10%10ml,10%20ml或25%10ml。肌注用25%溶液,每次10ml。或以此量加5%~10%溶液10ml,以5%葡萄糖液10ml稀释后缓慢静注。也可用门冬氨酸钾镁溶液,能同时补钾盐。
4.不良反应 镁为心脏及中枢神经的抑制剂,静脉给药或肌注可产生血压下降,甚至心脏停搏;可产生室性早搏,或室性心动过速。毒性心电图表现包括P-R延长,QRS增宽。血清镁浓度高于2mmol/L可出现高镁血症的症状:出汗、面部潮红、低血压、深腱反射抑制、弛缓性麻痹、体温降低、心功能抑制、中枢神经抑制,甚至导致致命性呼吸抑制。可产生低血钙。
中毒治疗为立即静注10%葡萄糖酸钙10~20ml。钙盐可迅速对抗镁对中枢神经系统的作用。
5.药物相互作用 于洋地黄化病人,当镁中毒给予钙时可产生传导阻滞。当与其神经肌肉阻滞剂合用,可产生过分的神经肌肉阻滞。
五、血管扩张剂、其他降压药、抗心绞痛药
近年在心血管急诊应用相当广泛的一类药物为血管扩张剂,常用于治疗高血压、急性和慢性心力衰竭、心绞痛、瓣膜疾病等。血管扩张剂根据其主要作用部位可区分为三大类:有些扩张动脉为主,直接松弛血管平滑肌,或激动血管壁α受体;有些主要扩张静脉;还有些均衡扩张动脉和静脉(表22-9)。
表22-9 常用血管扩张剂分类——根据其作用部位
| 动脉 | 静脉 | 混合性动脉/静脉 |
| 血管扩张剂 | 血管扩张剂 | 血管扩张剂 |
| 1.平滑肌松弛剂 | 硝酸盐类 | 1.α受体阻滞剂 |
| 肼苯达嗪 | 硝酸甘油 | 酚妥拉明 |
| 长压定 | 二硝酸异山梨醇酯 | 酚苄明 |
| 2.β[XB]2[/XB]受体激动剂 | 单硝酸异山梨醇酯 | 派唑嗪 |
| 沙丁胺醇 | 2.转化酶抑制剂 | |
| 对羟苯心胺 | 巯甲丙脯酸 | |
| 叔丁喘宁 | 苯酯丙脯酸 | |
| 3.硝普钠 | ||
| 4.钙拮抗剂 | ||
| 硝苯吡啶 |
高血压特征为周围血管阻力增加,直接作用于血管平滑肌的药物使阻力血管松弛而迅速降低血压。血管扩张剂治疗心力衰竭的作用机制一方面通过扩张静脉,增加静脉容量,减少回心静脉血量,降低肺毛细血管压和左室舒张末期压(前负荷),从而减轻肺充血;另一方面,也通过扩张小动脉,减轻外周血管阻力,从而减轻后负荷,改善左心室泵血功能,增加心搏量及心排血量,使左室舒张期容量减少,肺毛细血管压降低,从而心力衰竭好转。血管扩张剂减轻前、后负荷,也减少心肌氧消耗,有利改善心肌缺血(图22-2)。
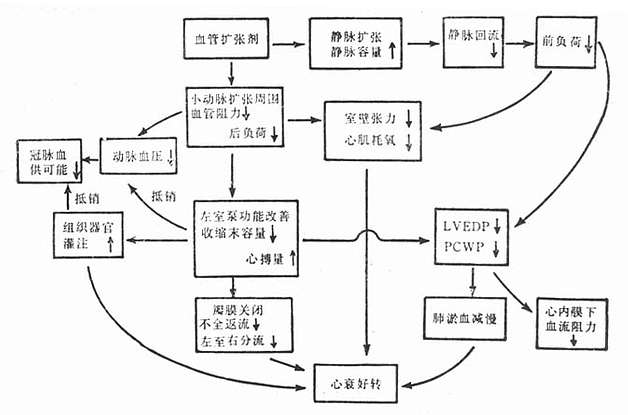
图22-2 血管扩张剂治疗心衰的机理
血管扩张剂能诱发或加剧严重心力衰竭患者的低血压,而使其应用受限制。血管扩张剂的效应取决其剂量。一般分三个阶段。小剂量时心排血量增加,肺毛血管压降低而动脉压很少变化;如滴注速度加快,在进一步增加心排血量及降低肺毛血管压同时,动脉压也开始下降;如再增快速度,则心排血量、肺毛血管压及动脉压均下降,严重时甚至可危及生命。因此多主张治疗心衰时采用小剂量以发挥其治疗效果和避免量过大。应用时必需遵循两项原则:①只用于伴肺充血、肺毛血管压力增高者,当后者低于2.0~2.4kPa(15~18mmHg)(临床估计,肺底无啰音),不再应用这类药物;②使动脉压保持在生理范围内,一般收缩压不应低于13.3kPa(100mmHg)。
(一)硝普钠(Nitroprusside sodium)
1.药理作用 为一种强力短效血管扩张剂,直接使小动脉及静脉平滑肌松弛,降低周围血管阻力及使静脉贮血。由于血压下降,可引起轻至中度心动过速,对心肌梗死和心力衰竭可改善心排血量。
2.药代学 仅静脉给药。1~2min内起效,作用持续5~10min。一分子硝普钠释出5分子氰化物离子,可与红细胞及组织的巯基结合,形成氰化物,后者于肝转化为硫氰盐。硝普钠半减期极短,约几秒钟;而硫氰盐半减期为7天,几全部经尿排泄。
3.适应证 各种高血压危象,需迅速降压,如高血压脑病、急进型高血压、急或慢性肾小球肾炎或急性左心衰竭合伴高血压急诊、颅内出血、嗜铬细胞瘤、主动脉夹层动脉瘤;急性心肌梗死时的心源性休克伴血管收缩状或左心衰竭;其他原因所致左心衰竭。
4.剂量、用法 硝普钠为水溶粉剂,每支50mg,用5%葡萄糖液溶解(其他溶液可能产生毒性产物)。为避免开始血压下降过快,宜先静点5%葡萄糖液,调节速度(8滴/min开始),再将新鲜溶好的25~50mg加入500ml溶液中。从15μg/min开始(伴高血压者可自25μg/min开始),每3~5min监测血压,调整滴速,直至肺毛血管楔嵌压降至正常,而动脉血压在13.3kPa(100mmHg)以上。如无血液动力学监测,在血压增高者,速度为25~400μg/min,血压正常者为25~150μg/min。对硝普钠的通常反应,血压立即下降30%~40%。对心衰病人,注意使血压下降不超过2.67kPa(20mmHg),心率增加不超过20次/min。
硝普钠水溶液不稳定,应临用前新鲜配制溶液。遇光易降解为铁氰化钠和氰化物。使溶液颜色变深褐,故滴注过程所用滴器,滴管均应避光(可用黑布、黑纸或银纸包遮)。治疗时间长,特别肾功能衰竭者应监测血硫氰盐浓度(毒性开始于50~100μg/ml)。
5.不良反应 滴注过快可产生恶心、呕吐、烦躁不安、头痛、头晕、胸骨后不适、腹痛、肌肉颤搐等;也可产生皮肤潮红、精神症状、减慢或中止滴注可迅速好转。用药时间长,硫氰盐蓄集产生中毒症状,如手足抽搐性痉挛、缺氧综合征。可引起进展迅速的甲状腺功能减退,明显乏力、恶心、低血压甚至死亡。可产生变性血红蛋白,维生素B[XB]12[/XB]缺乏,乳酸性酸中毒。硫氰盐可被透析消除。羟钴胺(Hyhroxycobalamine)也可用于治疗中毒。
(二)酚妥拉明(苄胺唑啉,Phentolamine,Regitine)
1.药理作用 为一种α受体阻滞剂。对突触前受体(α[XB]1[/XB])和突触后受体(α[XB]2[/XB])均有强的阻滞作用。对心肌有直接的正性肌力和变时性作用;对血管壁平滑肌还有直接松弛作用,可降血压。对胃肠道还有组胺样作用。
血液动力学效应似硝普钠,降低小动脉张力,也减低静脉张力,增加静脉容量,降低增高的左室舒张末压;降低动脉压,使降低的心排血量明显增高。易引起心动过速,加上正性肌力作用,可增加正肌氧耗量。静滴5min达作用高峰,作用持续15~30min。
2.治疗应用 适应证为预防和治疗嗜铬细胞瘤高血压危象(术前准备或手术时);服单胺氧化酶抑制病人的高血压危象;抗休克,当有过分血管收缩时,尤其当肺毛血管楔压大于2.67kPa(20mmHg)或中心静脉压大于1.96kPa(20cmH[XB]2[/XB]O);难治性心力衰竭;肾上腺素或多巴胺等缩血管药漏出血管外,可作皮肤浸润,以预防坏死;嗜铬细胞瘤诊断(酚妥拉明试验)。老年人、溃疡病患者禁用。
针剂每支10mg/1ml。10mg加入5%葡萄糖液50~100ml,以0.1~1mg/min速率静滴。口服每片25mg。
3.不良反应 静脉用药可引起窦性心动过速或快速性心律失常,心绞痛发作,头晕、恶心、面部潮红;严重者可有低血压;大剂量可产生鼻堵、腹泻、溃疡活动。
4,药物相互作用 可与去甲肾上腺素、多巴胺等合用,以治疗休克;可与心得安合用,治疗嗜铬细胞瘤高血压。宜待酚妥拉明作用过后应用洋地黄制剂。
(三)硝酸盐类(Nitrates)急诊常用的有硝酸甘油(Nitroglycerin,GTN),二硝酸异梨醇酯(消心痛,Isorbide nitrate,Sorbitrate,Isoket),单硝酸异梨醇酯(Isorbide-5-mononitrate,Elantan)。
1.药理作用 硝酸盐类松弛血管壁平滑肌,特别是毛细血管后的血管,包括大静脉。增加外周血池,减少回心血量,减轻左室舒张末压;也轻度降低动脉压力,扩张冠状动脉,有利减少心肌氧耗量;此外尚能促进侧支循环,增加缺血区的血流量,以缓解心绞痛,也用以治疗心力衰竭。
硝酸盐类对其他平滑肌也有松弛作用,可缓解幽门痉挛、胆绞痛、肾绞痛等。
2.药代学 硝酸甘油口服,因100%的首过肝代谢而无效。可自口腔粘膜和皮肤吸收,但也迅速被肝代谢。T[XB]1/2[/XB]1~2min。少数资料提示硝酸盐反覆应用可使循环效应产生耐受性(动物可发生)。骤停药可能导致血管收缩。
二硝酸异山梨醇酯口服后,20%~70%经肝“首过”代谢为单硝酸脂,后者也有扩血管作用。T[XB]1/2[/XB]:消心痛为0.5h;2-单硝酸异山梨醇酯为1.5h;5-单硝酸异山梨醇酯为5h(4~6h)。5-单硝酸代谢产物吸收慢,半减期长,可产生较恒定的效应,有异于硝酸甘油。由肾排泄的为原形的药物部分,为灭活的代谢产物。
3.适应证(静脉用药) 左心衰竭(尤其急性心肌梗死所致);急性心肌梗死伴发严重的心绞痛;严重心绞痛,口服抗心绞痛药效差;外科过程中产生可控制的低血压。
4.禁忌证 过敏体质(对硝酸盐类过敏史);低血压或未纠治的低血容量(可产生严重低血压,器官灌流不足,甚至血栓形成);肺毛细血管楔压正常或降低的患者;颅内压增高(如颅脑外伤、脑出血等);缩窄性心包炎或心包压塞。
5.用法、剂量 硝酸甘油片剂为每片0.5mg或0.6mg,舌下含;注射剂为1mg/1m1或50mg/10ml(Tridil)。一支(1mg)临用前以5%~10%葡萄糖液100~200ml稀释后静滴,开始剂量5~10μg/min,每隔5min可增加剂量,最大量不超过200μg/min。一般剂量0.6~12mg/h。有效后,应缓慢减量,不应骤停,防心绞痛复发。
经皮贴剂为每贴含硝酸甘油为0.5、5、10、15mg不等。每24h一贴,贴天胸腹部皮肤。
5-单硝酸异山梨醇酯(Elantan)片剂每片20mg或40mg。每次20mg~40mg,每日3次。
6.不良反应 低血压伴反射性心动过速,静脉用药时易有此作用,如血压降低明显,应暂中止滴注,抬高肢体,很快消失。出现正铁血红蛋白血症,当硝酸甘油剂量超过500μg/min,可致紫绀;正铁血红蛋白血浓度>1.5%,也可出现紫绀。用药过程如出现紫绀,应减量或中止滴注,必要时给予美蓝,2mg/kg,10min内静脉注射。头痛常见,此外可有心悸、心动过速、头晕、焦虑不安、胸骨后不适、肌肉搐搦、腹痛等。
7.药物相互作用 不宜与硝普钠等其他扩血管药合用;可与β受体阻滞剂或钙拮抗剂合用。静脉用药时,应停用其他硝酸盐类药物。
(四)巯甲丙脯酸(Captopril,Capotan)
1.药理作用 为一种转化酶(即激肽酶Ⅰ)竞争性口服抑制剂,抑制血管紧张素转化酶活性,降低血管紧张素Ⅱ(有强烈收缩血管作用),舒张小动脉;也抑制内源性缓激肽降解;抑制醛固酮形成和抗利尿激素分泌,对各类型高血压均有明显降压作用;对高肾素和正常肾素和正常肾素型高血压降压最明显,对低肾素型高血压加用利尿剂后降压也明显;由于扩张小动脉、小静脉,减轻心室前、后负荷,尤其是后负荷,从而改善心脏功能。
2.药代学 口服吸收迅速,1h达血浓度高峰,一般吸收70%,食物可降低药物吸收率至30%。60%以原型自尿排出,余下30%代谢为灭活性产物。蛋白结合率30%;T[XB]1/2[/XB]为4h。肾功能损害时蛋白结合减少,半减期延长。
3.治疗应用 适用于治疗常规疗法效差的严重高血压(各种类型);常规方法疗效差的慢性难治性心力衰竭。过敏体质者忌用。
每片为25mg、50mg或100mg。治疗高血压时每次25mg,每日3次(饭前服用);渐增量至每次50~100mg。每日最大量为450mg。治疗心力衰竭从小剂量6.25~12.5mg开始,每日3次。效差病例可加利尿剂。
4.不良反应 常用有皮疹、发热、关节痛、搔痒、味觉障碍、消化道症状;长程用药个别可发生蛋白尿、膜性肾小球病变或肾病综合征;粒细胞缺乏症、中性细胞减少,减量或停药后上述副反应可消失或避免。
(五)降压嗪(氯甲苯噻嗪唑,低压唑,Diazoxide,gyperstat)
1.药理作用 为一种非利尿性噻嗪类。直接松弛动脉平滑肌,降低周围血管阻力,使血压急剧下降。增快心率,反射性使心排血量增加。初始,肾血流量下降,以后增加。抑制胰脏β细胞分泌胰岛素,可产生暂时性血糖增高,通常12h内作用消失。可产生钠、水潴留。
2.药代学 分布容积0.2L/kg,与蛋白结合率为90%~95%。肾功能不全时结合率减少。本品可通过胎盘。T[XB]1/2[/XB]为20~40h(成人),55%~60%被代谢,其余尿排出。静脉注射,血压下降于1~5min最显著,随后10~20min血压相对迅速回升,降血压作用持续2~12h。口服100%吸收。
3.治疗应用 用于高血压危象(高血压脑病、急进型高血压),此外可作为升血糖药,治疗胰岛细胞瘤、糖元累积症、幼儿特发性低血糖症引起的低血糖。
禁用于嗜铬细胞瘤,与单胺氧化酶抑制剂反应有关的高血压危象,继发于主动脉缩窄或动静脉分流引起的高血压,合并心力衰竭、缺血性心脏病或糖尿病的高血压,对噻嗪类药物过敏者。妊娠、肾功能不全慎用。
针剂每支300mg,附专用溶剂20ml。治疗高血压危象,静注,单剂75~150mg(1~3mg/kg,最大量150mg),卧位快速静注或小壶入,心避免与血浆蛋白结合;必要时5min后重复1次剂量或静滴5mg/kg。
4.不良反应 常见的严重不良反应为浮肿、心衰加重(重复用药易有)、低血糖。不常见的严重不良反应为低血压伴心动过速,心绞痛,心肌梗死、尿素氮增高。可出现一时性脑缺血,发热感,头痛、恶心、失眠、便秘、腹部不适等。治疗严重低血压有时可用多巴胺,心动过速用心得安。
5.药
■[此处缺少一些内容]■
?技亮靠晌?.25mg,1~2h后如无过分血压下降可增量。
4.不良反应 一般剂量可引起鼻塞、乏力。较大剂量(>0.5mg/d)可引起嗜睡、体重增加、阳萎、腹泻等;可引起帕金森综合征、精神忧郁症、溃疡病活动出血;妊娠期应用可增加胎儿呼吸系统合并症。
5.药物相互作用 与洋地黄合用,可增加洋地黄毒性,洋地黄也增加利血平的心脏作用,可发生室性快速性心律失常或心动过缓。与奎尼丁合用可增加心肌抑制和降压作用。与利尿剂或其他降压药合用,可增强降压效应。利血平降压作用可被拟交感胺类(如麻黄碱或三环抗忧郁剂)所拮抗,胍乙啶减弱其降压作用和产生心动过缓或精神抑郁。麻醉药显著增强利血平降压作用,手术前两周应停本品,或诱导麻醉前预先用阿托品防止过度心动过缓。与单胺氧化酶抑制剂合用可突然血压升高或严重忧郁症。
六、抗凝药、溶栓药
(一)肝素
1.药理作用 是一种粘多糖硫酸酯,其结构中所含磺胺基有强负电荷,是抗凝血活性的有效基团。肝素有多方面的作用。
(1)抗凝血:对凝血链锁反应的多个环节,血小板粘附、聚集,以及纤维蛋白溶解系统均有影响。肝素的抗凝活性是由于它与蛋白质,尤其凝血因子相互作用而引起。肝素需抗凝血酶Ⅲ为辅因子,凝血酶及凝血因子Ⅸ、Ⅹ、Ⅺ等,都是具有以丝氨酸为活性中心的蛋白水解酶。肝素与抗凝血酶Ⅲ的赖氨酸残基结合,发生空间构象改变,使它易于与活性丝氨酸部位反应,因而抑制上述凝血因子的促凝活性,主要抑制凝血酶和激活的凝血因子Ⅹ。由于抑制了在凝血机制中占中心位置的激活的凝血因子Ⅹ,使凝血酶原不能转为凝血酶;由于也抑制已形成的凝血酶活性,使纤维蛋白原不能转变为纤维蛋白,使凝血时间延长。肝素也抑制凝血酶对Ⅷ、Ⅴ、Ⅷ因子的激活,血小板第3因子释放等方面的作用。肝素也抑制纤维蛋白所致的血小板聚集。
(2)激活脂蛋白脂肪酶:促使血脂蛋白转运,脂血廓清。
2.药代学 口服无效,必需注射。静滴部分被血小板第4因子中和。主要在肝代谢(80%),休克时肝血流量减少,代谢减弱。20%以原型尿排出。T[XB]1/2[/XB]依赖于剂量,小剂量时(<5000IU,静滴)约为1h;较大剂量时2~6h。不通过胎盘,不自乳汁排泄,可用于妊娠。
3.治疗应用 肝素抗凝作用迅速,半减期短,常用于短期内需抗凝或口服抗凝剂之前。
适应证为治疗和预防血栓形成和栓塞,如严重不稳定型心绞痛而预防心肌梗死;深静脉炎,肺栓塞,肢体动脉栓塞等;弥漫性血管内凝血;心脏外科手术体外循环。禁忌证有出血素质或凝血障碍患者,活动性溃疡病,新创口未愈者,产后或先兆流产者,休克,感染性心内膜炎,严重肝、肾功能不全及恶性高血压患者。糖尿病患者慎用,血小板明显减少患者应减量,酸中毒者应先纠正酸中毒。阿司匹林、链激酶等避免与肝素合用。
国内针剂每支12500IU(1ml相当100mg)。负荷量为1mg/kg(或5000IU),静注或加入5%~10%葡萄糖液或生理盐水100ml滴注,每分钟20~30滴。维持量用1000~1500IU/h,或者每6h静滴(最好用输液泵)0.5mg/kg。预防用低剂量可以5000IU深部皮下(腹壁或骼嵴)注射,每8~12h一次。不宜肌注,因可造成注射部位血肿。还必须根据病情、病期、有无出血症状及实验室监测随时调整剂量。具体剂量应个体化。
凝血时间(试管法)为实用监测方法,可床旁进行。给药前测,下次给药前1h重复,使凝血时间控制在正常值的2~3倍(20~30min),正常值为8~12min。活化部分凝血活酶时间(APTT),正常值约35s,做为剂量指标宜控制在正常值的1.5~2.5倍(即60~80s)。凝血时间大于30min,或APTT大于100s则表示已过量。改口服抗凝药时,应有3~5天重叠。
4.不良反应 自发性出血,为主要的并发症,往往因过量或存在禁忌证,宜监测以避免。血小板减少(可逆性),偶有过敏反应,有荨麻疹、鼻炎、结合膜炎、发热等。长期使用患者,可发生暂时性秃发、骨质疏松等。
当肝素引起出血症状,或存在禁忌,需迅速中止其作用时,可用鱼精蛋白(Protaminesulfate)中和,1%鱼精蛋白5ml可中和肝素50mg(缓慢静注)。如最后一次肝素注射已2h,鱼精蛋白用量可减半。如因心脏手术、血透析,需迅速中和肝素,鱼精蛋白量可为肝素的1.5~2倍。大剂量鱼精蛋白本身可引起出血、过敏反应等,甚至危及生命。
5.药物相互作用 阿司匹林及其他水杨酸制剂给予用肝素治疗的病人,可能引起出血。口服抗凝药轻度增加肝素作用。抗组胺药、洋地黄、奎尼丁、青霉素、四环素等抑制肝素的抗凝作用。
溶血栓治疗是指用药物(激活剂)激活纤维蛋白溶解系统,使血浆素原转变成大量血浆素,后者引起纤维蛋白降解,从而加速已形成的血栓溶解。溶血栓治疗可使血块迅速、完全地溶解,但出血并发症发生率也较高。目前用于临床的主要有链激酶、尿激酶,尚在研究中的组织型血浆素原激活剂(TPA)将成为有前途的药物。
(二)链激酶(Streptokinase,SK)
1.药理作用 为一种间接的血浆素原激活剂(由β-溶血性链球菌所产生的非酶蛋白质)。它与人血浆素原或血浆素形成复合物后具有活性。循环复合物渗入血栓,并激活吸附、沉积在血栓中的血浆素原,引起血栓内在性溶解;循环中所产生的血浆素和血浆中的抗血浆素结合,在血栓形成部位释放,产生血栓的外源性溶解。SK是一种异体蛋白,可以引起免疫反应而降低药物效应,并引起过敏反应。既往曾有链球菌感染的患者,体内几乎均存在抗链球菌激酶抗体,故首次剂量必须超过溶栓的需要量以中和这种抗体。
2.药代学 T[XB]1/2[/XB]为10~12min。
3.适应证 肺栓塞;深部静脉血栓形成;周围动脉栓塞;急性心肌梗死,症状开始6h内;动静脉瘘血栓形成(血透析病人)。
4,禁忌证 绝对禁忌证为活动性内出血;新近发生的脑血管意外、脑肿瘤(2个月内),中度的凝血机制障碍、血小板缺陷或出血素质。相对禁忌证为外科大手术、创伤,或创伤性检查后未满10天;未控制的严重高血压;心房颤动(可能促发脑栓塞);妊娠或产后未满10天;心、肺复苏后伴肋骨骨折;原有内科出血条件者,如支气管扩张,严重的肝、肾功能不全,脓毒性血栓静脉炎。
5.用法、剂量 开始治疗前应测定凝血时间、凝血酶时间、活化部分凝血激酶时间、凝血酶原时间、血红蛋白、血小板计数、血球积压,以除外凝血障碍及作为以后监测的基准。
粉剂每瓶100000或600000IU。静滴时150万IU,60min内持续滴注,监测上述指标以调整剂量,使凝血酶时间延长至正常的2~4倍。开始治疗前如已用肝素或口服抗凝药,需等APTT或凝血酶原时间恢复后再开始本药治疗,用本药后需再用肝素治疗以预防血栓形成再发。
6.不良反应
(1)出血:为最常见的并发症。严重者需中止治疗,发生率约25%。尽量减少静脉穿刺次数,避免肌肉注射或动脉取标本,不同时使用非类固醇抗类药或口服抗凝药,以减少出血的发生。出血处理,一般终止滴注,溶解活性即可中止。如出血严重,应输鲜血,或新鲜冰冻血浆,有时需用氨已酸(EACA)或氨甲苯酸(PAMBA)以拮抗血浆素原。如静脉穿刺部位持续渗血可用浸有EACA的止血棉按压止血。
(2)过敏反应:用SK治疗者发生率约15%。包括荨麻疹、潮红、头痛,偶发生支气管痉挛、低血压。可用氢化考的松静脉注射(100mg)。为预防其发生,可给药前30min肌注非那更25mg,或静注地塞米松2.5~5mg。
(三)尿激素(Urokinase,UK)
1.药理作用 是一种由肾细胞合成的蛋白水解酶,为血浆素原的直接激活剂、人肾组织培养中含有大量以无活力的前体形式存在的尿激酶,通过蛋白水解作用而形成具活力的尿激酶,低分子系列可作为临床溶栓药。人尿提取的高、低分子系列可作为溶栓药、尿激酶直接使血浆素原转变为血浆素,对纤维蛋白原或其他血浆蛋白有溶解作用。
2.药代学 T[XB]1/2[/XB]为12~16min.
3.治疗应用 适应证、禁忌证均同SK。粉针剂每瓶分别为6000、60000、250000IU等。负荷量为40万IU/min,静脉注射(10min),以后以40~60万IU于1~2h内静滴。UK目前价格昂贵。
4. 不良反应 也可引起出血、发热,但罕引起过敏反应,此点不同于SK。
(四)组织血浆素原激活剂(Tissueplasminogen actirator,t-PA)
为一种血浆素原的直接激活剂,与纤维蛋白结合的亲和力很强,大大超过尿激素。t-PA与血栓内的纤维蛋白结合成复合体,作用于血浆素原转变为血浆素而溶解新鲜的纤维蛋白,t-PA只引起局部的溶纤而不产生全身溶栓状态,此为前述两者所不及。t-PA主要来自血管壁,由内皮细胞合成,并不断释放。1983年自人黑色素细胞瘤细胞株中找出携带t-PA的基因,通过DNA重组技术,在大肠杆菌中进行表达后,能大量生产t-PA。t-PA不具抗原性,不会引起过敏反应,生物半减期短暂,有利于溶栓失败者及时进行冠状动脉旁路搭桥和冠状动脉形成术,为很有前途的溶栓药物。治疗应用的适应证同SK。剂量10mg于10min内静注;继以第1h输入50mg,第2h40mg。
(游凯)
参考文献
[1] Pentel P,et al: Drug therapyfor cardiac emergencies:pharmacokinetic consideration.In: scheinman MM(ed):Cardiac Emergencies.p288,Saunders. philadelphia. 1984
[2] Goldberger E(ed):Teatment ofCardiac Emergencies 4th ed.p323,Mosby,St.Louis. 1985
[3] Leier CV(ed):CardiotonicDrugs.A clinical Survey pp85,199,Marcell Dekker.New York. 1982
[4] Ewy GA,Bressler R(eds):Cardiovascular Drugs And the Management of HeartDisease.p57,69,95,103,157.Raven. New York.1982
[5] Conh CR(ed): Cardiac DrugTherapy.Davis.philadelphia.1984
[6] Gould LA(ed:) Drug Treatmentof Cardiac Arrhythmias.Futura.Mount Kisco.1983
[7] 陈新谦等(主编):新编药物学,第12版。人民卫生出版社,1985
[8] Bochner F et al(eds):Handbook of Clinical Pharmacology.2nd ed .Little Brown.Boston.1983
[9] British National FormularyNo.9 London.1985
[10] Gillies Hc,et al(eds):ATexbook of Clinical pharmacology.2nd ed. Hodder and Stoughton.London 1986
[11] Gilman AG,Goodman LS,etal(eds): The pharmacological Basis of the Therapeutics.7th edp716,1338.MacMillan.New York ,1985
[12] Laffel GL et al:Thrombolytic Therapy : a new strategyfor the treatment of acute myocardial infarction.N Eng1 J Med 1984; 311:710,770